



SHOPPING CART
Your cart is empty.
shop now
Your cart is empty.
shop now
DSC stands for Differential Scanning Calorimeter Analysis, which measures the relationship between the temperature or time and the difference in heat flow or power between the sample and reference material. It can be used to measure various properties of solid and liquid materials, including melting point, boiling point, glass transition, heat capacity, crystallization temperature, crystallinity, purity, reaction temperature, and reaction heat, including those of polymer materials.
The predecessor of DSC is DTA (Differential Thermal Analysis). The difference between DSC and DTA measurement principle is that DSC maintains △T = 0 and measures the relationship between △H-T under the control of temperature change, while DTA provides the same heat to the sample and reference to measure the relationship between △T-T. The biggest difference between DSC and DTA is that DTA can only be qualitative or semi-quantitative, while DSC can be quantitatively analyzed.
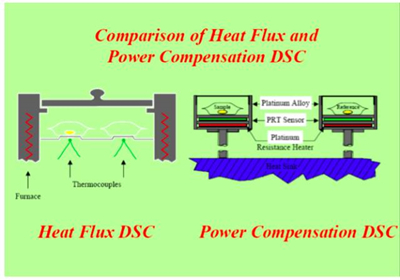
DSC curve: the vertical axis is the difference in heat rate (dQ/dt) between the sample and the reference material, in units of milliwatts (mW); the horizontal axis is temperature or time. The DSC spectrum must indicate the direction of the endothermic and exothermic effects.
1. Essence of DSC Y-Axis
(1)When the sample quality remains unchanged and there is no reaction, the vertical coordinate is the heat capacity Cp.
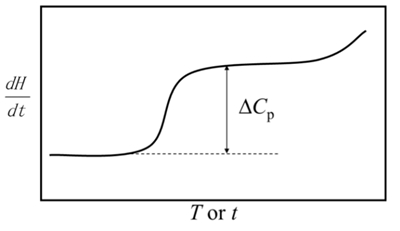
(2)When reaction occurs: the curve peaks; the area of the peak=reaction enthalpy+enthalpy of heat capacity change, where the enthalpy of heat capacity change is often neglected.
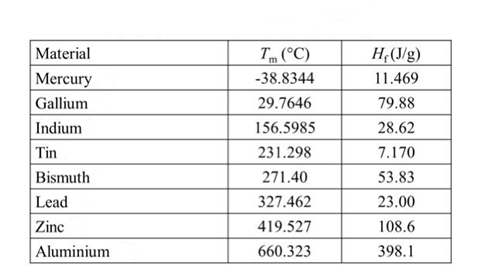
Calibrate the temperature and heat simultaneously using a calibration material. The calibration material should be highly pure (≧99.999%), have known physical characteristics, be non-hygroscopic, stable to light, non-decomposable, non-toxic, non-reactive to the vessel or atmosphere, and non-flammable or explosive. Before calibration, the vessel should be thoroughly cleaned to ensure that the calibration material has no adsorption layer or oxidation layer, and be accurately weighed. The standard materials recommended by the International Thermal Analysis and Calorimetry Association include cyclopentane, water, indium, benzoic acid, tin, aluminum, etc.
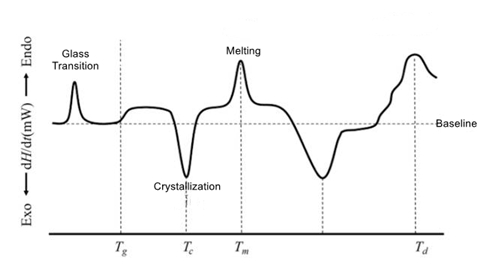
2. Glass Transition and Enthalpy Relaxation
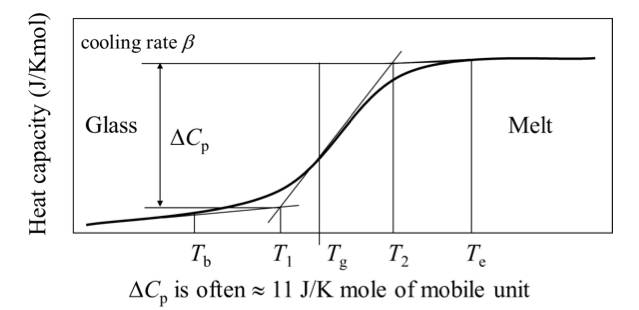
(1) The faster the cooling rate, the higher the glassy state thermal capacity.
(2) When the system is annealed at a certain temperature Ta in the glassy state, the heat capacity decreases with the annealing time, and the enthalpy also decreases, which is called thermal enthalpy relaxation. Among them, a = Tg-Ta is called the overcooling of annealing.
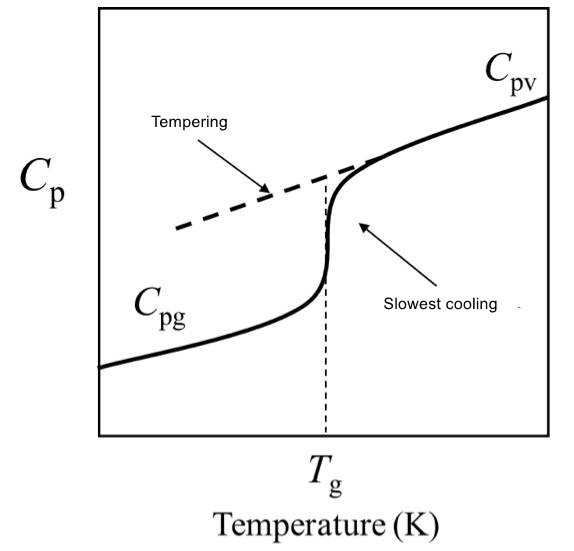
(3) The maximum possible relaxed enthalpy (infinite annealing time) of a sample in the tempered glass state is referred to as the excess enthalpy, denoted as △H0.
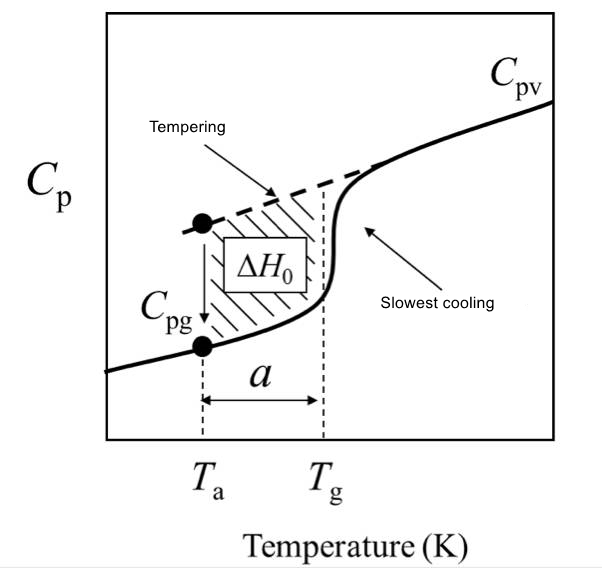
(4) The excess enthalpy of relaxation can be compensated when heated, which can be measured by DSC.
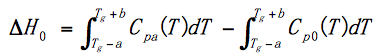
Cp0(T) is a thermal capacity curve without heat enthalpy compensation.
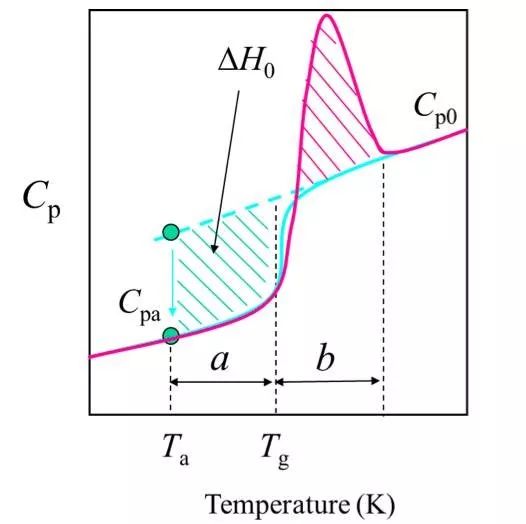
(5) If the annealing time t is limited, the relaxation enthalpy is △Ha <△H0.
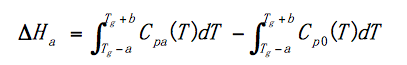
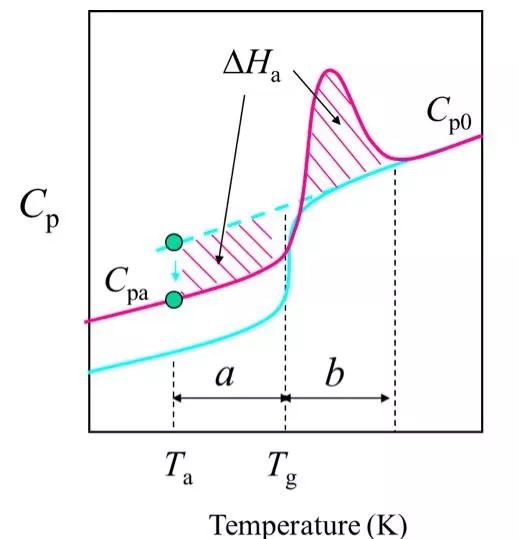
(6) Relaxation excess enthalpy △Ha is a function of time, the longer the annealing time, the larger the relaxation enthalpy, and more enthalpy needs to be compensated.
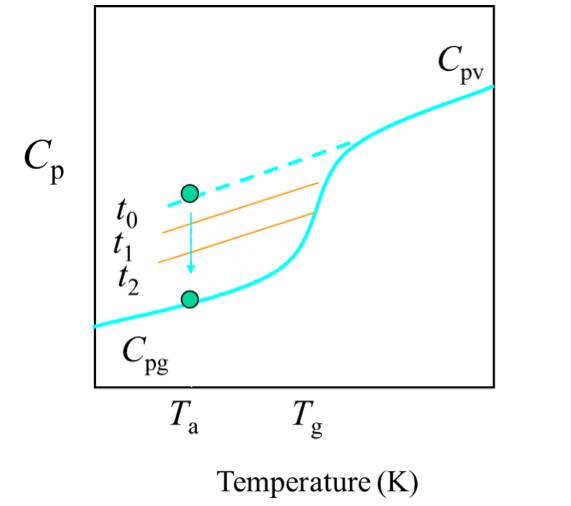
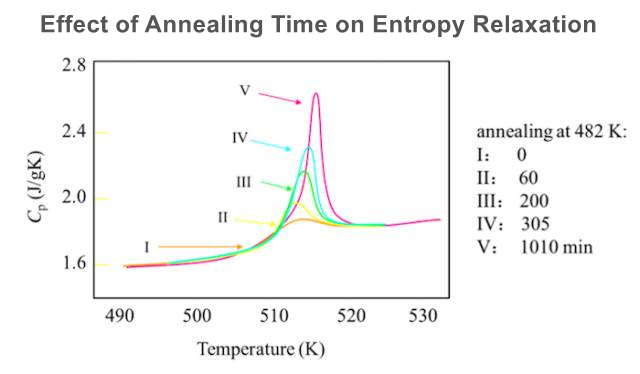
(7) Sample size 10-15 mg; Heat at 20 °C / min to a temperature above the thermal relaxation enthalpy, eliminate thermal history; Reduce the temperature to 50 °C below the estimated Tg at the fastest rate; Then heat at 20 °C / min to determine Tg; Compare the sample weight before and after the measurement. If weight loss is found, repeat the above process.
3. Melting and Crystallization
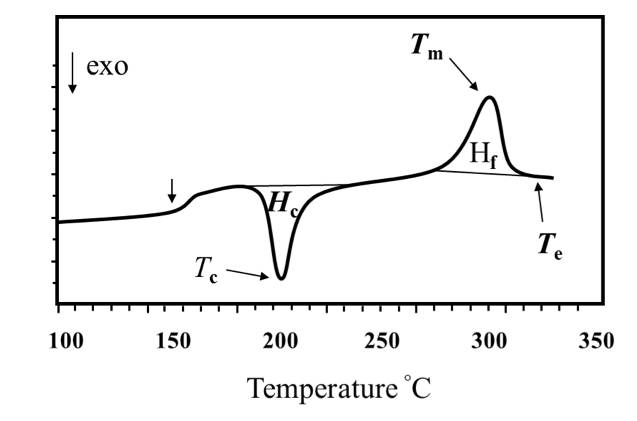
Three parameters representing melting: [1] Tm: the peak value of endothermic heat; [2]) Hf: the area of the endothermic peak; [3]Te: the complete melting temperature.
Two parameters representing the crystallization: [1] Tc: the peak value of heat release; [2] Hc: the area of heat release peak.
(1) The relationship between sample quantity and Tm value:
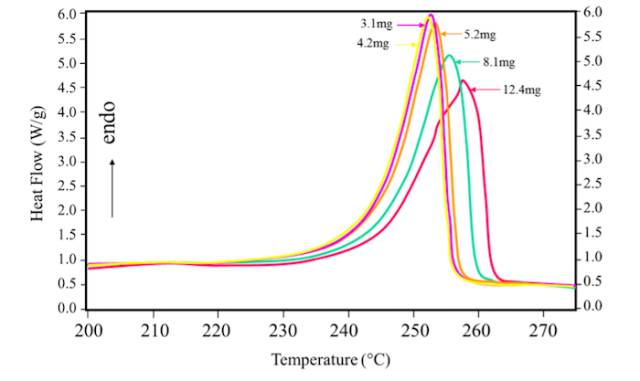
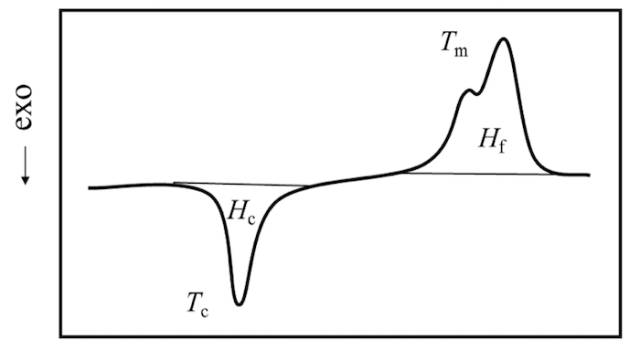
Melt peak has width, called melt limit; often out of the shoulder, even a double peak; melting point is always higher than the crystallization temperature. Reasons could be summarized as follow:
a, crystallization and melting are not reciprocal processes;
b, melting point, crystallization temperature and wafer thickness are related;
c, wafer is in non-equilibrium state and wafer spontaneously thickens.
Thompson-Gibbs Formula:
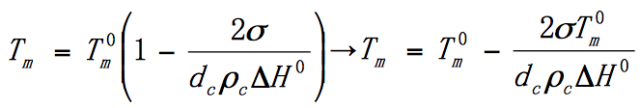
The relationship between melting point and chip thickness is that the thicker the chip, the higher the melting point. And the infinitely thick chip, the equilibrium melting point.
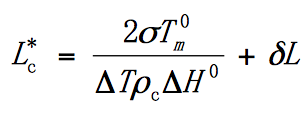
The relationship between the crystallization temperature and the chip thickness is that the higher the crystallization temperature, the thicker the initial chip. And the chip is unbalanced and has a thickening tendency.
In conclusion, the chip is thickened, so the melting point is definitely higher than the crystallization temperature, the thicker the thickness, the higher the melting point.The crystallization and melting points must be cycled repeatedly with heating or cooling to obtain repeatable data. The repeatability of Tm and Tc measurements is around ±3°C, which is higher than Tg measurements.


C082.Wangdefu International Building, Furong, Changsha, Hunan, China


+86-19907415020





